În anul 2008, Spitalul Județean de Urgență Buzău a încheiat contract pentru derularea Programului Național de Tratament al Hemofiliei și Talasemiei. Coordonatorul acestui program este Dr. Anica Gina - medic specialist Pediatrie. Beneficiarii acestui program sunt atât copii cât și adulți. În România, sunt înregistraţi în evidenţele specialiştilor aproximativ 2.000 de pacienţi cu hemofilie şi boala von Willebrand.
Ce este Hemofilia?
Hemofilia este o boală înnăscută și moștenită de la mamă, care are la baza deficitul unui factor ce participă la închegarea sângelui: factorul (F) VIII sau IX. în aproximativ 30% dintre cazuri, boala este sporadică, cu istoric familial negativ. Hemofilia este o boală cronică (durează întreaga viață, necesitând un tratament de suplimentare cu factorul deficient permanent), în care este afectată formarea cheagului de sânge sau coagularea sângelui. Cheagul de sânge apare acolo unde peretele unui vas de sânge, mai mare sau mai mic, a fost deteriorat, tăiat sau rupt.
Hemofilia este o afecțiune rară, frecvența ei în populație se situează în jur de 100/1 milion de locuitori, hemofilia A fiind de aproximativ 5-6 ori mai frecventă decât hemofilia B.
Caracteristic pentru această boală este faptul ca afectează de o manieră simptomatică doar pe cei de sex masculin; rareori (sub 30-40% dintre cazuri) pot apărea sângerări excesive la femeile purtătoare sau conductoare.
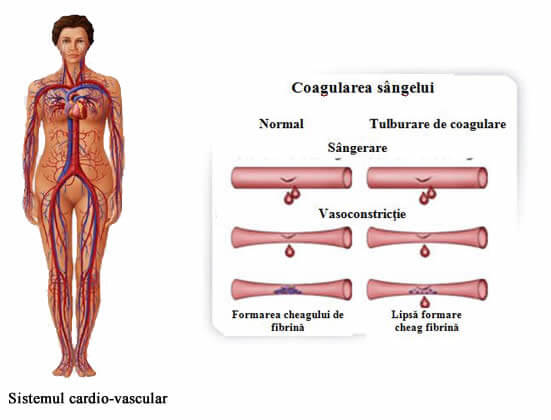
Nota definitorie a bolii este sângerarea excesivă, prelungită, fără tendință de sistare. Aceste vase de sânge sunt asemenea unor tuburi elastice prin care circulă sângele. Dacă ne tăiem, ne lovim sau cădem, peretele subțire al vasului de sânge se poate rupe.
Pentru a „repara” ruptura, sângele aduce în zona afectată trombocite și un număr de structuri (proteine), care au fost notate și denumite pentru a ne ajuta să înțelegem ce se întâmplă - factori - și au fost numerotate de la I la XIII. Aceștia formează împreună o rețea, un dop sau un cheag oprind astfel ieșirea sângelui în afara vasului de sânge. În cazul în care unul dintre acești factori lipsește complet sau parțial sau nu funcționează adecvat, acest proces de coagulare nu se poate realiza în timp util, rezultând un grup de boli numite tulburări de coagulare, dintre care face parte atât hemofilia, cât și boala înrudită, von Willebrand.
Toate tipurile de hemofilie se caracterizează prin tendința la sângerare sau hemoragie. Indiferent de sediul unde se produce sângerarea, în lipsa unui tratament corespunzător cu factorul de coagulare care lipsește din organism, pierderea de sânge poate deveni foarte gravă.
În funcție de factorul de coagulare care lipsește, hemofilia se clasifică astfel:
- hemofilie A - în care lipsește factorul de coagulare VIII;
- hemofilie B - în care lipsește factorul de coagulare IX;
- hemofilie C - în care lipsește factorul de coagulare XI.
Riscul de a se produce o hemoragie la pacienții cu hemofilie și gravitatea acesteia depinde de cantitatea de factor de coagulare prezentă în sânge. După cantitatea de factor de coagulare prezentă în sânge hemofilia se clasifică în:
- forma severă - în sânge există sub 1 % din cantitatea normală de factor de coagulare;
- forma medie - în sânge este prezentă între 1-4% din cantitatea de factor normală;
- forma ușoară - pacientul are 5-30% din cantitatea normală de factor de coagulare ce caracterizează o persoană sănătoasă.
De gravitatea bolii depinde gradul de risc pentru hemoragii al pacientului. În general, o formă gravă va fi diagnosticată în primii 2 ani de viață, când copilul începe să umble, cade și se lovește des, forma medie puțin mai târziu, iar forma ușoară poate fi pusă în evidență uneori numai la vârsta de adult, cu ocazia unor accidente sau intervenții chirurgicale.
Totuși, forma ușoară nu trebuie considerată lipsită de importanță. Pacientul cunoscut cu forma ușoară va informa medicul cu ocazia unei extracții dentare sau intervenții chirurgicale asupra bolii sale, pentru ca acesta să adopte o conduită corespunzătoare, riscul sângerărilor provocate fiind real. Există și excepții în care formele severe de deficit au un comportament benign. Aceste situații sunt foarte rare (sub 10%), adevărate excepții.
Tratament
Tratamentul pentru persoanele diagnosticate cu hemofilie este asigurat prin programul naţional de profil. Principala activitate a acestui program este asigurarea, în spital şi în ambulatoriu, prin farmaciile cu circuit închis, a medicamentelor specifice pentru prevenţia şi tratamentul accidentelor hemoragice ale bolnavilor cu hemofilie congenitală (hemofilia A şi B, boala von Willebrand) şi hemofilie dobândită.
Tratamentul hemofiliei se stabilește în funcție de severitatea bolii. Întrucât hemofilia este o boală genetică, tratamentul se începe de cele mai multe ori de la naștere. Hemofilia se tratează, în principiu, prin înlocuirea factorilor coagulării anormali pentru a preveni pierderile severe de sânge și complicațiile sângerării.
La nivelul Spitalului Județean de Urgență Buzău, prin acest program se asigură tratamentul profilactic pentru copiii diagnosticați cu hemofilie, tratamentul episoadelor de sângerare acută a tuturor pacienților (adulți și copii), recuperare medicală prin kinetoterapie.
- Sursa:
- Asociația Română de Hemofilie
- Irish Haemofilia Society
- Casa Județeană de Asigurări de Sănătate Buzău
Adriana Bunilă